




材料AI迈向「可解释时代」,日本团队破解高维光谱黑箱,锁定新材料发现关键特征
超神经HyperAI热度: 4473
日本东京科学研究所团队提出可解释深度学习方法,利用ALIGNN模型和层次聚类分析,从2681种无机晶体的高维光吸收光谱数据中提取关键结构特征(如V/Cr元素及四面体配位环境),实现光谱预测与物理解释的统一,推动材料AI进入可解释时代。
 摘要由 Mars AI 生成
摘要由 Mars AI 生成本摘要由 Mars AI 模型生成,其生成内容的准确性、完整性还处于迭代更新阶段。
来自日本东京科学研究所的研究团队提出了一种深度学习模型的解释方法,能够处理材料科学中的高维光谱数据。研究人员构建了一个包含 2681 种氧化物、硫族化合物及相关化合物的光吸收光谱第一性原理计算数据集。与标准密度泛函计算相比,对光谱起始能量和形状进行校正后,计算结果与已报道的实验光谱吻合度显著提高。
近年来,机器学习在材料科学领域的应用备受关注,其应用已从早期的结构–性质标量预测(如带隙能量、点缺陷形成能、熔点等)逐步扩展到更复杂的高维物理量建模,其中最具挑战性的方向之一便是材料光谱的预测与解析。
介电函数、光谱(吸收、反射和发射)以及电子和声子态密度等光谱数据对于理解和设计材料至关重要。然而,与标量性质相比,高维光谱数据具有输出维度大、结构复杂、物理约束强等特点,使得传统机器学习方法难以同时兼顾精度与可解释性。深度学习模型虽然已经能够在一定程度上实现对光谱的预测,但「可解释性缺失」仍然是制约其在材料设计中进一步应用的关键瓶颈。
在此背景下,来自日本东京科学研究所的研究团队提出了一种深度学习模型的解释方法,能够处理材料科学中的高维光谱数据。研究人员构建了一个包含 2,681 种氧化物、硫族化合物及相关化合物的光吸收光谱第一性原理计算数据集。与标准密度泛函计算相比,对光谱起始能量和形状进行校正后,计算结果与已报道的实验光谱吻合度显著提高。
研究人员还利用该数据集和 ALIGNN 算法开发了一个高精度的光吸收光谱预测模型。通过特征提取和聚类分析相结合的方法,成功提取了主要决定光吸收起始能量和强度的关键元素种类及其配位环境。
相关研究成果以「Deep Learning–Based Extraction of Promising Material Groups and Common Features from High-Dimensional Data: A Case of Optical Spectra of Inorganic Crystals」为题,发表于 Advanced Intelligent Discovery。
研究亮点:
* 本研究提出了一种通过高维光谱数据的特征提取与聚类分析进行材料分类,从而提取潜在材料群组及其共性特征的方法。
* 本研究构建的第一性原理计算数据集和机器学习模型有望在未来的材料发现和材料信息学研究中发挥重要作用。
* 本研究提出的方法具有广泛的适用性,可用于各种光谱数据的分类和解释,其应用范围不仅限于无机晶体的光吸收光谱。

论文地址:https://advanced.onlinelibrary.wiley.com/doi/10.1002/aidi.202600007
利用高通量第一性原理计算构建数据集
研究人员首先从 Materials Project 数据库中筛选满足以下条件的氧化物、硫族化合物及相关材料:(1)材料至少包含 O、S、Se 中的一种元素,且其氧化数不一定为 −2;(2)材料不包含以下元素:H、卤素、稀有气体、Mn–Ni、Tc–Rh、Os–Ir、Po、镧系元素(La 与 Ce 除外)以及锕系元素;(3)材料不表现出自旋极化;(4)由于计算成本过高或晶体结构存在不确定性,排除空间群为 P1 且/或原始晶胞中原子数超过 40 的体系。
最终用于第一性原理计算的材料总数为 9,808 种,并按照下图所示流程构建计算数据库。
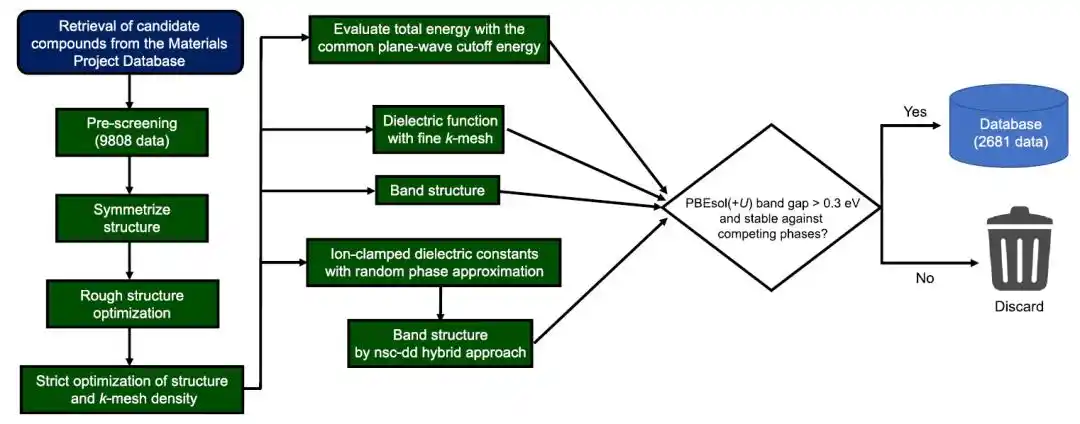
构建金属氧化物、硫族化合物及相关化合物介电函数的第一性原理计算数据库的工作流程
如图所示,该计算流程极为复杂,为了在保持一致性与高效利用计算资源的同时进行高通量计算,研究人员使用自研程序,并依赖 pymatgen、FireWorks、Custodian、atomate 以及 vise 等工具完成流程自动化。所有第一性原理计算均基于 VASP 软件包完成。该工作流程基于 PBEsol(+U) 计算生成光学吸收光谱与化合物形成能,并基于 nsc-dd 混合泛函以及 PBEsol(+U) 计算得到能带结构。
在机器学习数据集方面,研究人员剔除了:(1)在本地数据库中相对于竞争相不稳定的材料;(2)PBEsol(+U)带隙小于 0.3 eV 的材料。最终保留下来的材料数量为2681种。
构建基于光吸收光谱的 ALIGNN 模型
机器学习模型构建及预测精度
在模型层面,本研究采用 ALIGNN(Atomistic Line Graph Neural Network)作为核心预测框架,用于建模高维光学吸收谱。与传统晶体图卷积网络(CGCNN)相比,ALIGNN 的核心优势在于同时引入「原子图 + 键线图」双重表示,从而显式编码三体角度信息,实现对局域结构环境更精细的表达,下图上半部分给出了 ALIGNN 架构的示意图。
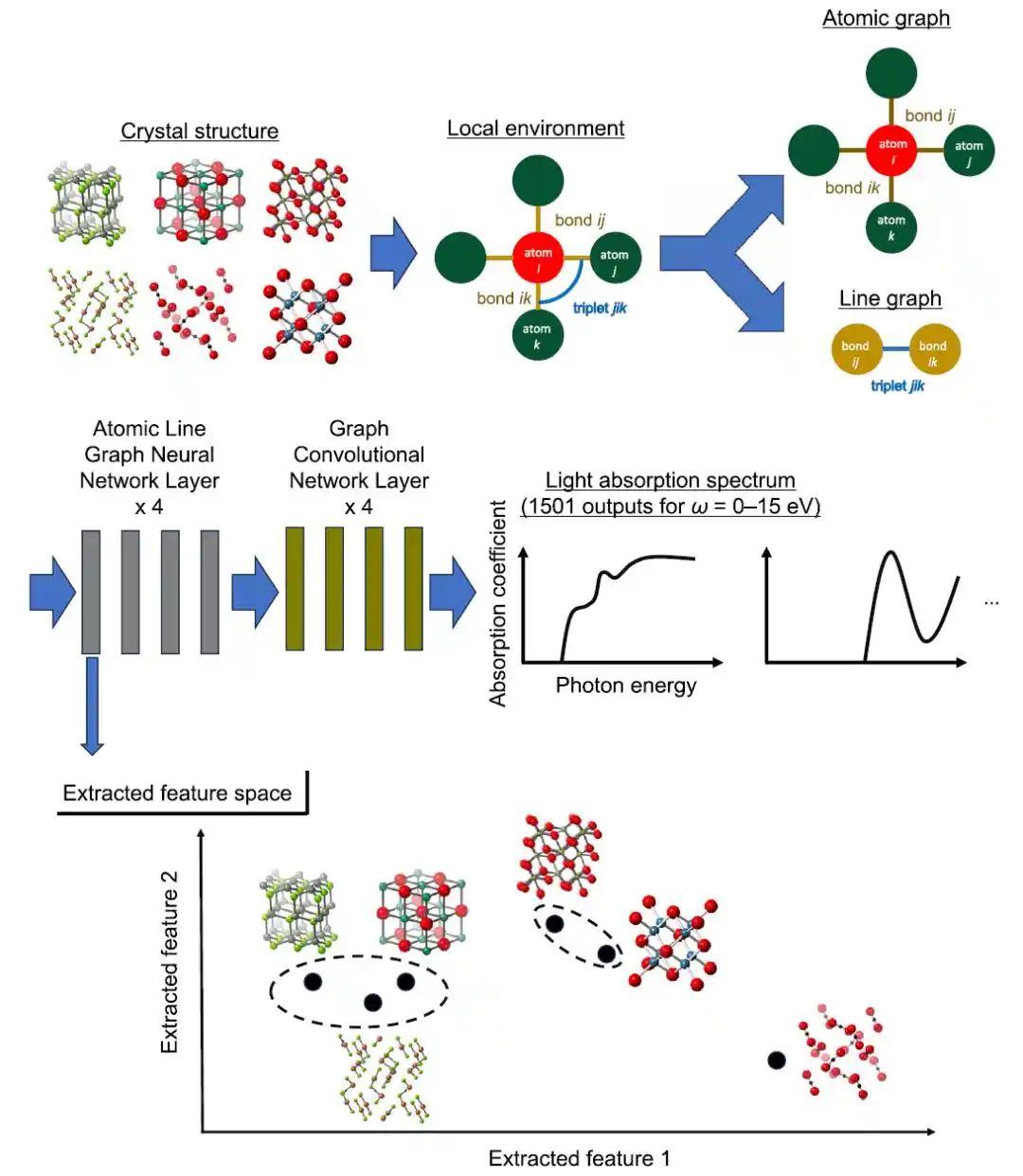
用于光学吸收光谱预测的ALIGNN模型及所提出解释方法的整体示意图
在该框架中,原子作为节点,原子间键作为边,并进一步将「边之间的关系」构建为线图,从而将键角信息转化为可学习的结构特征。这一设计使模型不仅捕捉两体距离信息,还能够刻画三体相互作用,从而更贴近真实的晶体物理行为。
特征提取和聚类
研究人员对优化模型的第一层 ALIGNN 进行了特征提取,并对每种材料所有原子位点的特征向量进行平均,然后进行层次聚类分析,如上图下半部分所示。该方法的目标是将材料分类为在输入特征(例如元素组成和原子配位特征,包括相邻原子数、原子间距离和键角)和输出属性(光吸收光谱)方面均表现出相似性的组。
下图显示了通过层次聚类得到的 96 个组的光学吸收光谱,每个簇内的光谱形状确实相似,证实了本研究聚类方法的有效性。

通过层次聚类得到的吸收光谱分类结果
成果:实现了材料群体结构与物理机制的可解释性提取
研究人员为了验证新深度学习模型在处理材料科学中高维光谱数据的能力,进行了系列试验:
预测性能能力
在预测性能方面,ALIGNN 模型在测试集中整体表现出较高精度,如下图所示,其中约 75% 的材料吸收谱预测平均绝对误差(MAE)低于 0.14,说明模型能够较好重现复杂光谱形状。
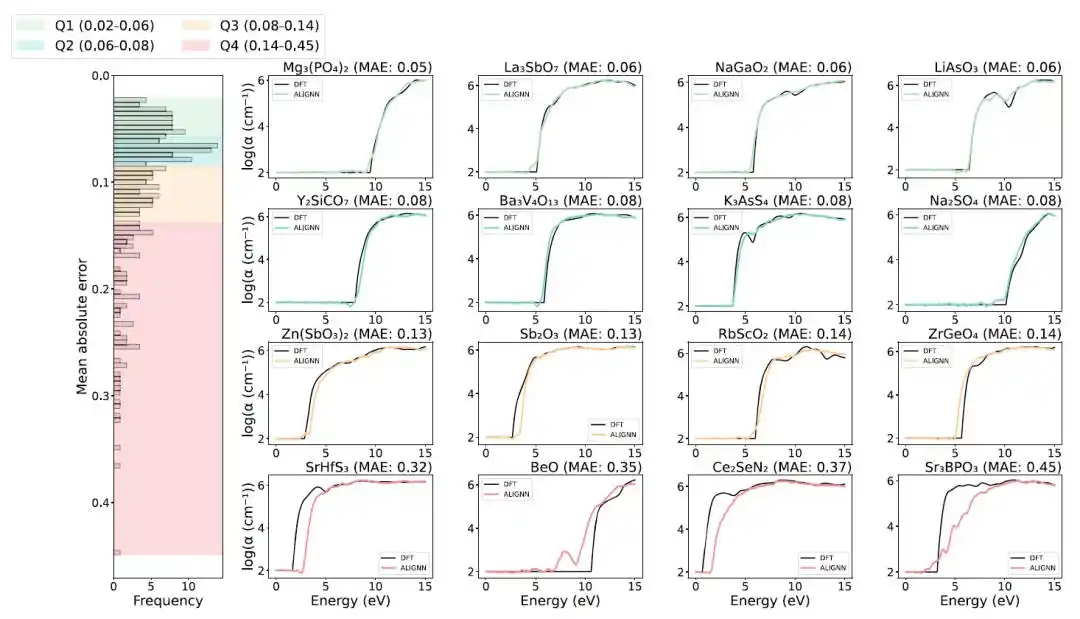
使用优化后的ALIGNN模型对测试集光学吸收光谱的预测结果
上图右侧面板展示了在各四分位区间中误差最大的四种材料的预测结果。对于前三个四分位区间中的材料,ALIGNN 预测结果(彩色曲线)与第一性原理参考计算结果(黑色曲线)吻合良;然而,第四四分位中的部分化合物在光学吸收光谱的起始位置上出现明显偏差,这些离群样本预测性能较差,主要原因在于其独特的电子结构,以及训练数据集中相似结构材料的不足。
捕捉光学吸收光谱起始位置的能力
尽管 MAE 是一个覆盖整个光谱范围的全局指标,研究人员还进一步考察了模型是否能够准确再现局部光谱起始能量。下图给出了一个一致性(parity)图:比较第一性原理计算与 ALIGNN 预测中,当 log₁₀ α(ω) 首次超过 2.5 时对应的最低光子能量,其中 α 表示吸收系数。
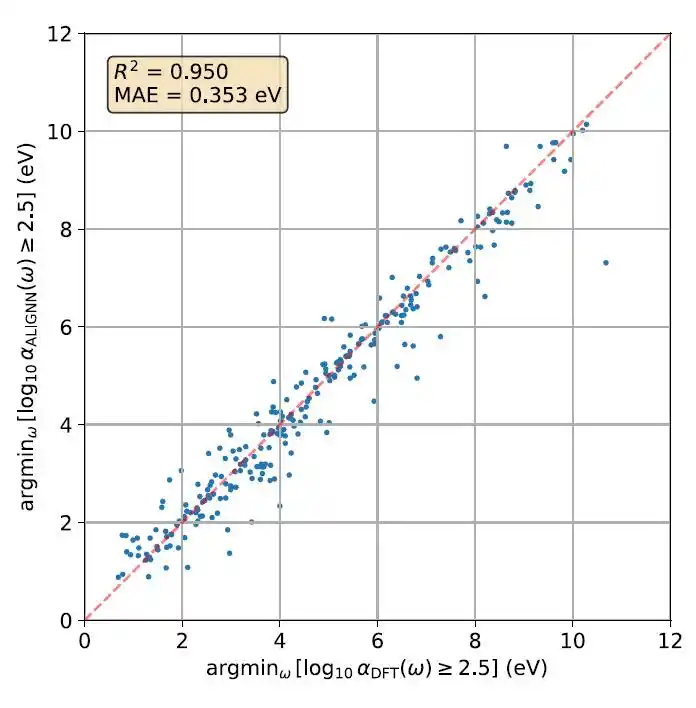
测试集光谱起始能量的配对图(parity plot)
结果显示,该起始能量预测的 R² 为 0.950,MAE 为 0.353 eV,表明 ALIGNN 模型能够较为准确地捕捉光学吸收光谱的起始位置。
可解释性分析
在可解释性分析方面,研究人员从 ALIGNN 第一层提取特征表示,并对材料进行层次聚类,从而形成 96 个材料群组。结果显示,同一簇内材料在光谱形状上高度一致,尤其在吸收起始位置与吸收边陡峭程度方面表现出明显共性,这说明模型在早期层已学习到与光谱相关的结构特征表达。
而进一步的案例分析显示,不同材料群组具有清晰的物理差异。以第 74 簇为例,该簇中的材料通常具有较宽的带隙以及在光谱起始附近较高的吸收系数。下图 a 显示,该簇中的所有材料均包含 V 或 Cr 元素,其他阳离子则主要为碱金属,这些材料大多以 VO₄³⁻、CrO₄²⁻ 或 Cr₂O₇²⁻ 形式存在,其中阳离子处于四面体配位环境。
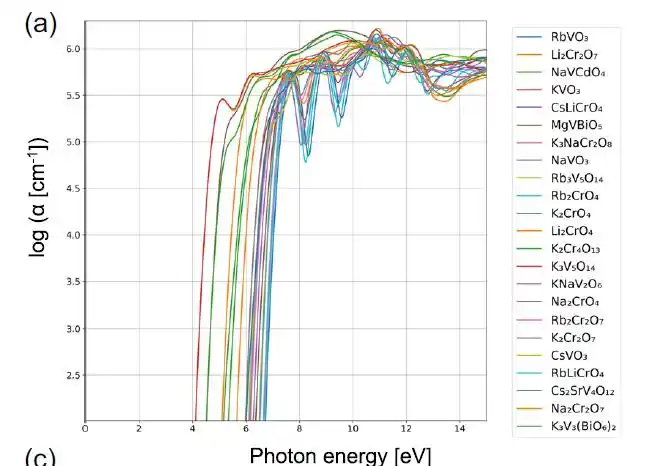
属于第 74 簇的物质的光学吸收光谱,其中 α 表示吸收系数
研究人员使用 matminer 中实现的 CrystalFingerprintNN,对该簇中各材料的阳离子位点四面体配位指数进行计算,并分析所有阳离子位点最大值的分布。如下图 b 所示,大多数材料确实存在四面体配位位点。
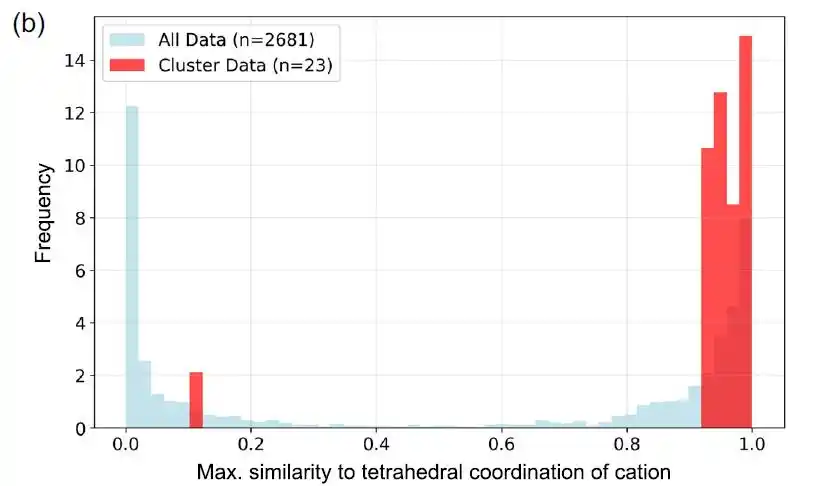
第 74 簇材料(红色)与整体数据集(蓝色)的四面体配位相似性分布
从电子态密度来看,在导带底(CBM)附近可以观察到由 V-d 或 Cr-d 态引起的尖锐峰值。V⁵⁺ 与 Cr⁶⁺ 的高价态提供了大量可用于光学跃迁的未占据电子态。因此,从固体化学与物理角度来看,这些钒酸盐、铬酸盐与重铬酸盐具有较高的光学吸收系数是合理的。
这种从「模型聚类结果」反推「化学机制」的过程,使得机器学习结果不再是黑箱预测,而成为可用于材料设计的知识来源。此外,研究还对比了基于原始光谱数据直接聚类的结果,发现其虽然能够识别相似光谱,但难以形成清晰的化学结构分组,材料类型混杂严重。这进一步证明 ALIGNN 特征空间在「结构–性质一致性表达」方面的优势。
结语
本研究的意义不仅在于构建了一个高精度光学吸收光谱预测模型,更重要的是提出了一种将「深度学习表示学习」与「材料物理解释」相结合的方法框架。通过 ALIGNN 模型与层次聚类分析的结合,研究实现了从高维光谱数据中提取材料共性规律的能力,使得机器学习模型不仅能够预测结果,还能够揭示结果背后的结构与电子起源。
理想情况下,需要将电子–空穴相互作用、电子–声子耦合以及点缺陷的影响纳入考虑,以分别再现激子效应、声子辅助电子跃迁以及缺陷相关的光谱特征。然而,在包含这些效应的情况下进行高通量第一性原理光谱计算计算成本过高,因此本研究未予实现。即便如此,未来随着更高精度多体计算方法与机器学习模型的进一步融合,该类研究有望在材料发现过程中发挥更加核心的作用,推动材料设计从经验驱动走向数据驱动与机制驱动融合的新阶段。
参考文献:https://advanced.onlinelibrary.wiley.com/doi/10.1002/aidi.202600007https://phys.org/news/2026-06-ai-materials-discovery-uncovering.html
本文来自微信公众号“HyperAI超神经”,作者:HyperAI
本内容旨在传递行业动态,不构成投资建议或承诺。



24H热门新闻
暂无内容